




Although relatively rare, the overall incidence of lysosomal storage is about 1 in every 5,000 live births. In addition, of the nearly 70 known lysosomal storage disorders, 70% affect the central nervous system. These single-gene disorders cause lysosomal dysfunction, resulting in metabolic instability, dysregulation of the mammalian target protein of rapamycin (mTOR, which normally inhibits inflammation), impaired autophagy, and nerve cell death. Several therapies targeting the underlying pathologic mechanisms of lysosomal storage disease have been approved or are under development, including enzyme replacement therapy, substrate reduction therapy, molecular chaperone therapy, gene therapy, gene editing, and neuroprotective therapy
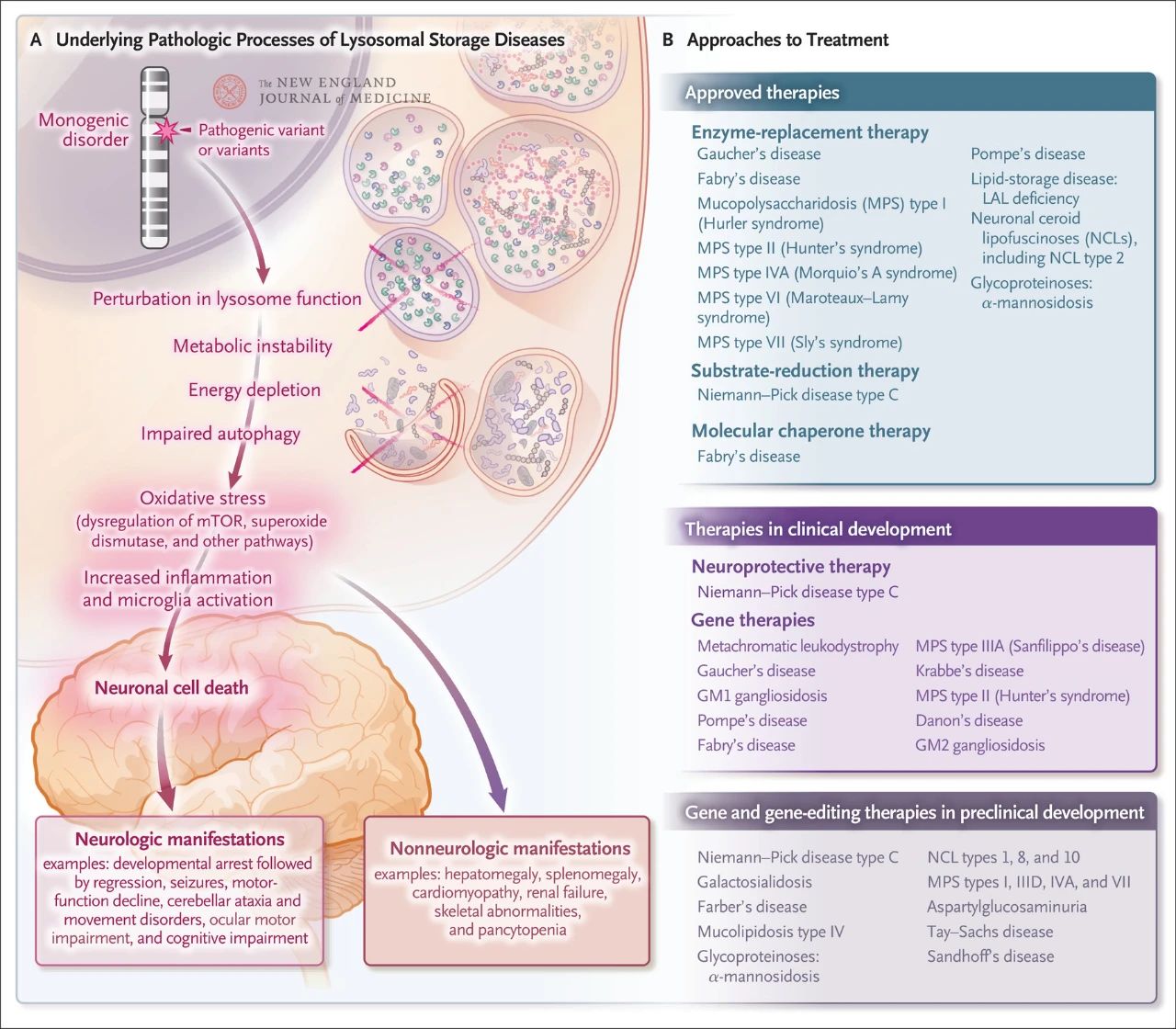
Niemann-pick disease type C is a lysosomal storage cellular cholesterol transport disorder caused by biallelic mutations in either NPC1 (95%) or NPC2 (5%). The symptoms of type C of Niemann-Pick disease include rapid, fatal neurological decline in infancy, while the late juvenile, juvenile, and adult onset forms include splenomegaly, supranuclear gaze paralysis and cerebellar ataxia, dysarticulationia, and progressive dementia.
In this issue of the journal, Bremova-Ertl et al report the results of a double-blind, placebo-controlled, crossover trial. The trial used a potential neuroprotective agent, the amino acid analogue N-acetyl-L-leucine (NALL), to treat Niemann-Pick disease type C. They recruited 60 symptomatic adolescent and adult patients and the results showed significant improvement in the total score (primary endpoint) of the Ataxia Assessment and Rating Scale.
The clinical trials of N-acetyl-DL-leucine (Tanganil), a racemic of NALL and n-acetyl-D-leucine, seem to be largely driven by experience: the mechanism of action has not been clearly elucidated. N-acetyl-dl-leucine has been approved for the treatment of acute vertigo since the 1950s; Animal models suggest that the drug works by rebalancing the overpolarization and depolarization of medial vestibular neurons. Subsequently, Strupp et al. reported the results of a short-term study in which they observed improvements in symptoms in 13 patients with degenerative cerebellar ataxia of various etiologies, findings that reignited interest in looking at the drug again.
The mechanism by which n-acetyl-DL-leucine improves nerve function is not yet clear, but the findings in two mouse models, one of Niemann-Pick disease type C and the other of GM2 ganglioside storage disorder Variant O (Sandhoff disease), another neurodegenerative lysosomal disease, have prompted attention to turn to NALL. Specifically, survival of Npc1-/- mice treated with n-acetyl-DL-leucine or NALL (L-enantiomers) improved, while survival of mice treated with n-acetyl-D-leucine (D-enantiomers) did not, suggesting that NALL is the active form of the drug. In a similar study of GM2 ganglioside storage disorder variant O (Hexb-/-), n-acetyl-DL-leucine resulted in a modest but significant extension of lifespan in mice.
To explore the mechanism of action of n-acetyl-DL-leucine, the researchers investigated the metabolic pathway of leucine by measuring metabolites in the cerebellar tissues of the mutant animals. In a variant O model of GM2 ganglioside storage disorder, n-acetyl-DL-leucine normalizes glucose and glutamate metabolism, increases autophagy, and increases levels of superoxide dismutase (an active oxygen scavger). In the C model of Niemann-Pick disease, changes in glucose and antioxidant metabolism and improvements in mitochondrial energy metabolism were observed. Although L-leucine is a potent mTOR activator, there was no change in the level or phosphorylation of mTOR after treatment with n-acetyl-DL-leucine or its enantiomers in either mouse model.
The neuroprotective effect of NALL has been observed in a mouse model of cortical impingement induced brain injury. These effects include lowering neuroinflammatory markers, reducing cortical cell death, and improving autophagy flux. After NALL treatment, the motor and cognitive functions of the injured mice were restored and the lesion size was reduced.
The inflammatory response of the central nervous system is the hallmark of most neurodegenerative lysosomal storage disorders. If neuroinflammation can be reduced with NALL treatment, the clinical symptoms of many, if not all, neurodegenerative lysosomal storage disorders may be improved. As this study shows, NALL is also expected to have synergies with other therapies for lysosomal storage disease.
Many lysosomal storage disorders are also associated with cerebellar ataxia. According to an international study involving children and adults with GM2 ganglioside storage disorders (Tay-Sachs disease and Sandhoff disease), ataxia was reduced and fine motor coordination improved after NALL treatment. However, a large, multicenter, double-blind, randomized, placebo-controlled trial showed that n-acetyl-DL-leucine was not clinically effective in patients with mixed (inherited, non-inherited, and unexplained) cerebellar ataxia. This finding suggests that efficacy may only be observed in trials involving patients with inherited cerebellar ataxia and the associated mechanisms of action analyzed. In addition, because NALL reduces neuroinflammation, which can lead to traumatic brain injury, trials of NALL for the treatment of traumatic brain injury may be considered.
Post time: Mar-02-2024